FASTQA-JS
To better enable researchers to analyze their own fastq files without downloading additional software, I created FASTQA-JS. Base quality scores often drop off towards the end of a read when errors accumulate in the spots of DNA on a flowcell. It is common practice to analyze these quality scores and truncate the reads when quality scores are below a specified threshold. Below is an example of the output from FASTQA-JS.
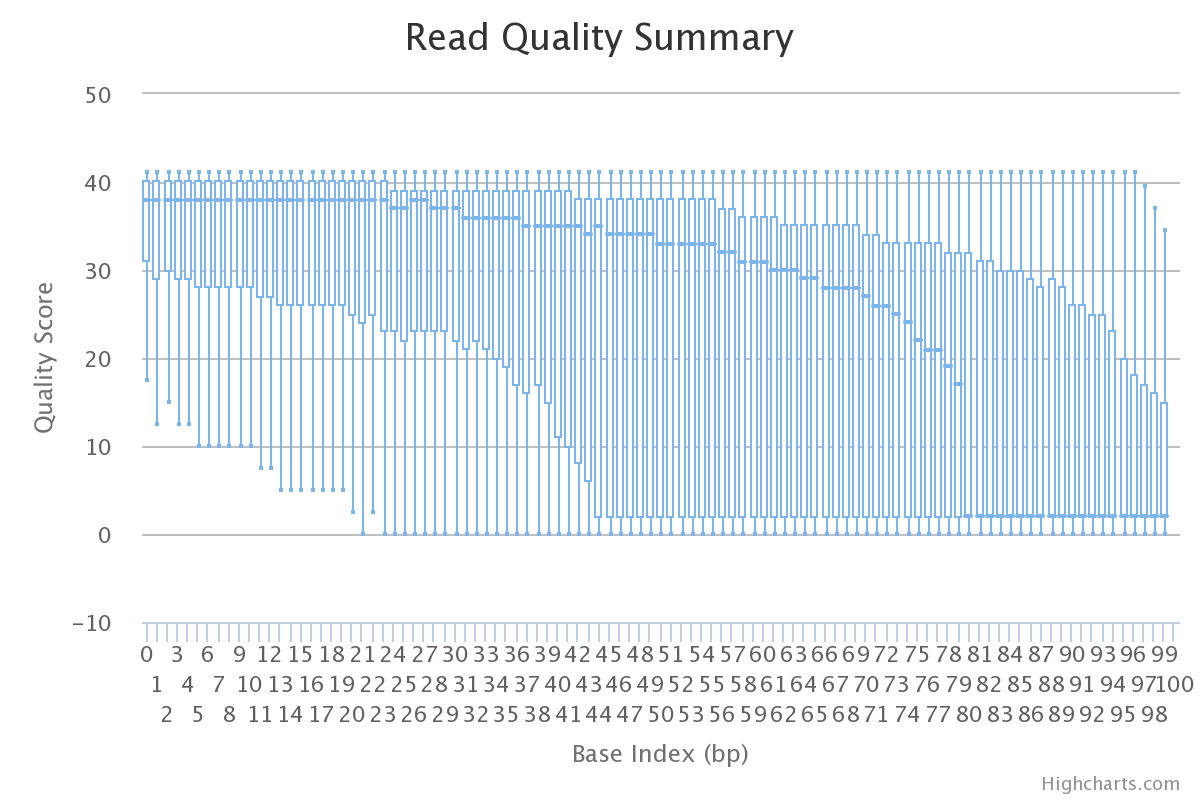
FASTQA-JS is built completely in JavaScript and will work on most modern browsers. Besides allowing for analysis without software installation, it also processes the fastq file on the client’s machine. By being processed in the browser on a client’s machine, their large fastq file never leaves their computer and they don’t have to wait on upload time nor managing remote file system space. This also led to a problem on my end because JavaScript is built to read entire files and a typical fastq file will easily max out the system memory of a common computer. I ended up relying on the FileReader api and individually processing sliced blobs of the file. I originally tried to do this in a nice for-loop, but the file I/O happens asynchronously from the rest of the JavaScript, so there was no way for me to trigger events dependent on the whole file to be read. I circumvented this problem by chaining the blob processing functions together, where they would either call the next chunk to be processed or call the “done” function, which would then format the data for highcharts.
I still have some more improvements to make with the tool, but I’m happy with the level of functionality I achieved and also surprised by JavaScript’s performance. This learning experience has been valuable and I plan on adding other statistics to the output in the future.
29 May 2014